Pārskats
Fenilketonūrija (fen-ul-keetone-YU-ree-ah jeb PKU) ir iedzimts vielmaiņas traucējums, kurā organisms nespēj pilnībā sadalīt olbaltumvielu (aminoskābi) fenilalanīnu. Tas notiek tāpēc, ka nepieciešamajam fermentam, fenilalanīna hidroksilāzei, ir deficīts. Tāpēc fenilalanīns uzkrājas ķermeņa šūnās un izraisa nervu sistēmu un smadzeņu bojājumus.
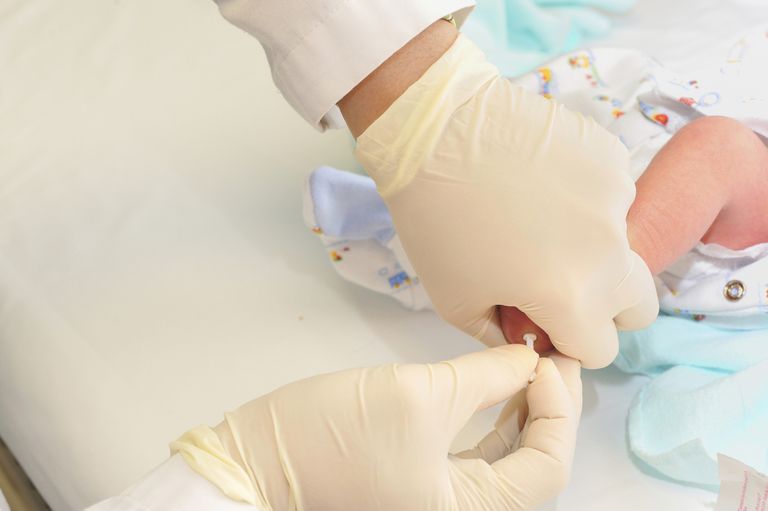
Fenilketonūrija ir ārstējama slimība, kuru var viegli noteikt ar vienkāršu asins analīzi. Amerikas Savienotajās Valstīs visiem jaundzimušajiem zīdaiņiem ir jāpārbauda PKU kā metabolisma un ģenētiskās skrīninga daļa, kas tiek veikta visiem jaundzimušajiem. Tiek pārbaudīti arī visi jaundzimušie Apvienotajā Karalistē, Kanādā, Austrālijā, Jaunzēlandē, Japānā, Rietumu un lielākajā daļā Austrumeiropas valstīs un daudzās citās valstīs visā pasaulē.
(Pārskats par PKU priekšlaicīgi dzimušiem bērniem ir atšķirīgs un grūtāks vairāku iemeslu dēļ.)
Katru gadu 10 000 līdz 15 000 zīdaiņu dzimst ar šo slimību Amerikas Savienotajās Valstīs, un fenilketonūrija notiek gan visu etnisko izcelsmi, gan vīriešiem un sievietēm (lai gan tas ir biežāk indivīdiem no Ziemeļeiropas un Native American mantojuma.)
Simptomi
Zīdainis, kas dzimuši ar fenilketonūriju, parasti attīstīsies pirmajos mēnešos. Ja neārstē, simptomi sāk attīstīties trīs līdz sešu mēnešu vecumā un var ietvert:
- novēlota attīstība
- garīgās paildzināšanās
- krampji
- Ļoti sausa āda, ekzēma un izsitumi
- izteikta "mousy" vai "musty" smarža urīns, elpa un sviedri
- viegls sejas, gaismas vai blondu mati
- aizkaitināmība, nemiers, hiperaktivitāte
diagnoze
fenilketonūrija tiek diagnosticēta ar asins analīžu palīdzību, parasti kā daļa no parastajiem skrīninga testiem, kas nodoti jaundzimušajam pirmajās dzīves dienās .
Ja klāt ir PKU, tad fenilalanīna līmenis asinīs būs augstāks nekā parasti.
Pārbaude ir ļoti precīza, ja to dara, ja bērns ir vecāks par 24 stundām, bet ir vecāks par septiņām dienām. Ja zīdaiņu testē vecāki par 24 stundām, ieteicams atkārtot testu, ja zīdainis ir vienu nedēļu vecs. Kā minēts iepriekš, pirmsdzemdību bērniem vajadzēja pārbaudīt citā veidā vairāku iemeslu dēļ, piemēram, lopbarības kavēšanās.
Ārstēšana
Tā kā fenilketonūrija ir fenilalanīna sadalīšanās problēma, zīdaiņiem tiek piešķirts īpašs diētu, kas fenilalanīnam ir ļoti zems.
Vispirms tiek izmantota īpaša zemu fenilalanīna zīdaiņu formula (Lofenalac).
Kad bērns aug, zāles ar zemu fenilalanīna saturu tiek pievienotas uzturam, bet nav atļauts lietot tādus proteīnus saturošus pārtikas produktus kā piens, olas, gaļa vai zivis. Maisīgais saldinātājs aspartāms (NutraSweet, Equal) satur fenilalanīnu, tādēļ tiek novērsti arī diētas dzērieni un pārtikas produkti, kas satur aspartāmu. Jūs, iespējams, atzīmējāt vietu, kurā atrodas bezalkoholiskie dzērieni, piemēram, diētika Kokss, kas norāda, ka produkts nedrīkst lietot cilvēki ar PKU. Bērniem un pusaudžiem indivīdiem jāpaliek ierobežotai ar fenilalanīnu.
Daži indivīdi spēj samazināt uztura ierobežojumus, kad viņi veci.
Lai noteiktu fenilalanīna līmeni, ir vajadzīgi regulāri asins analīzes, un, ja līmenis ir pārāk augsts, var būt nepieciešams pielāgot diētu. Papildus ierobežotajam diētam daži indivīdi var lietot narkotiku Kuvan (sapropterīns), lai palīdzētu samazināt fenilalanīna līmeni asinīs.
Uzraudzība
Kā atzīmēts, asins analīzes tiek izmantotas, lai uzraudzītu cilvēkus ar PKU. Pašlaik pamatnostādnēs ir ieteikts, ka fenilalanīna mērķa koncentrācijai asinīs jābūt no 120 līdz 360 μM cilvēkiem ar visu vecumu PKU. Dažreiz vecākiem pieaugušajiem ir pieļaujams maksimāli 600 μM. Grūtniecēm tomēr ir stingrāk jāievēro viņu uzturs, un ieteicamais maksimālais līmenis ir 240 μM.
Pētījumi par atbilstību (to cilvēku skaits, kuri ievēro viņu diētu un ievēro šos norādījumus) ir 88 procenti bērniem no dzimšanas līdz četriem gadiem, bet tikai 33 procenti tiem, kas ir vecumā no 30 gadiem.
Ģenētikas loma ─ PKU ir ģenētiskais traucējums, kas tiek nodots no vecākiem uz bērniem. Lai iegūtu PKU, bērnam ir jāpārmanto specifiska gūna mutācija PKU no
katram
vecākam. Ja bērns manto gēnu tikai no viena vecāka, tad bērnam ir arī PKU gēnu mutācija, bet tai nav PKU. Tie, kas manto tikai vienu gēnu mutāciju, nepārstrādā PKU, bet var nodot stāvokli saviem bērniem (būt par nesēju). Ja diviem vecākiem ir gēns, viņiem ir aptuveni 25 procentu iespēja, ka bērnam ir PKU, 25 procentiem izredzes, ka viņu bērns neizaugs PKU vai būs pārvadātājs, un 50 procentiem izredzes, ka viņu bērns būs arī slimības nesējs. Kad PKU tiek diagnosticēts mazulim, tad šim bērnam ir jāievēro PKU ēdienu plāns visā savas dzīves laikā.
PKU grūtniecības laikā
Jaunas sievietes ar fenilketonūriju, kuri neēd fenilalnīnu ierobežotu diētu, tad, kad iestājas grūtniecība, viņiem būs augsts fenilalanīna līmenis. Tas var radīt nopietnas medicīniskas problēmas, kas pazīstamas kā PKU sindroms bērnam, ieskaitot garīgo atpalicību, zemu dzimšanas svaru, dzemdību defektus sirdī vai citus iedzimtus defektus. Tomēr, ja jauna sieviete vismaz 3 mēnešus pirms grūtniecības perioda atsāk zemu fenilalanīna diētu un turpina uzturēties visā grūtniecības laikā, var novērst PKU sindromu. Citiem vārdiem sakot, sievietēm ar PKU ir iespējama veselīga grūtniecība, ja vien viņi plāno uz priekšu un rūpīgi pārrauga diētu visā grūtniecības laikā.
Pētījumi
Pētnieki meklē veidus, kā izlabot fenilketonūriju, piemēram, aizstāt ar traucējumiem atbildīgo defektu gēnu vai izveidot ģenētiski modificētu enzīmu, lai aizstātu deficītu. Zinātnieki arī pētina ķīmiskus savienojumus, piemēram, tetrahidrobiopterīnu (BH4) un lielas neitrālās aminoskābes, kā līdzekļus PKU ārstēšanai, pazeminot fenilalanīna līmeni asinīs.
Pārvarēšana
Cīņa ar PKU ir sarežģīta un prasa lielu apņemšanos, jo tā ir mūža ilgums. Atbalsts var būt noderīgs, un ir daudz atbalsta grupu un atbalsta kopienas, kurās cilvēki var mijiedarboties ar citiem, kas strādā ar PKU gan emocionālajam atbalstam, gan jaunāko pētījumu laikā.
